K-mer
El terme k-mer fa referència a totes les subcadenes possibles de longitud k contingudes en una cadena. En genòmica computacional, els k-mers són les subsequències possibles (de longitud k) d'una lectura obtinguda a patir d'un procés de seqüenciació de l'ADN. La quantitat de k-mers possibles donada una cadena de longitud L és , mentre que el nombre de possibles k-mers donades n possibilitats (4 en el cas d'ADN, p. ex. ACTG) és . Els k-mers s'utilitzen normalment en l'assemblatge de seqüències, però també en l'alineament de seqüències.[1] En el context del genoma humà, k-mers de diverses longituds s'han utilitzat per explicar la variabilitat dels índexs de mutació.[2][3]


Assemblatge de seqüències
Resum
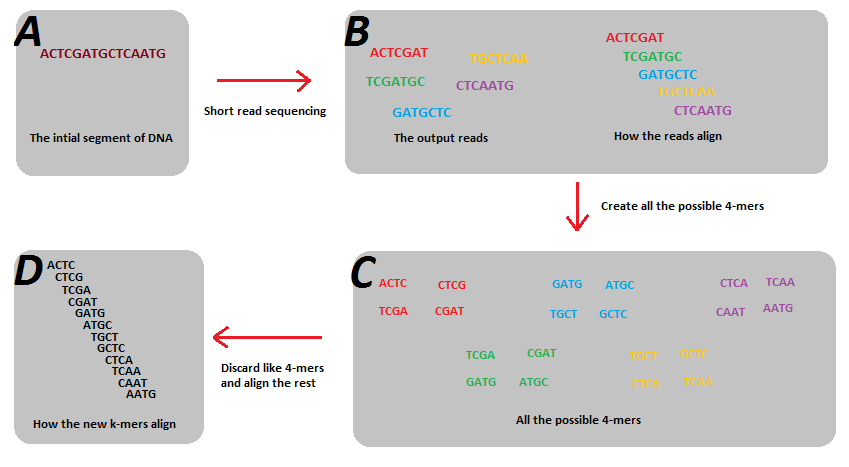
En el context de l'assemblatge de seqüències, els k-mers s'utilitzen en el procés de creació de grafs de Bruijn. Per tal de crear-ne un, les cadenes que s'emmagatzemen en cada aresta amb una longitud, , han de solapar-se amb una altra cadena en una altra aresta per per tal de crear un vertex. Les lectures obtingudes del procés de seqüenciació tindran diferents longituds. Posem el cas, amb la seqüenciació d'Illumina es poden generar 100-mers. Tanmateix, el problema amb la seqüenciació és que només fraccions petites dels possibles 100-mers presents en el genoma són generats al final. Això és a causa dels errors de lectura, però sobretot, simplement buits de cobertura que ocorren durant la seqüenciació. El problema és tanmateix, que aquestes fraccions més petites dels possible k-mers viola la suposició clau dels grafs de Bruijn que totes les lectures de k-mer han de sobreposar-se els seus k-mers adjunts en el genoma per (que no pot passar quan tots els k-mers no són presents). La solució a aquest problema és trencar aquestes lectures de mides del k-mer a una mida més petita que la dels k-mers, per tal que els k-mers més petits resultants representin tots el possibles k-mers d'aquella mida més petita que són present al genoma.[1] A més, partint el k-mers a mides més petites també ajudes a alleugerir el problema de diferents longituds de lectura inicials. Un exemple de solució de partir les lectures a k-mers més petits és mostra a la figura 1. En aquest exemple les 5 lectures no compten tot els possibles 7-mers del genoma, i com a tal, no pot crear-se un graf de Bruijn. Però quan es parteixi a 4-mers, les subcadenes resultants són suficients per a reconstruir el genoma que utilitza un graf de Bruijn.



Elecció de k-mer
L'elecció de la mida del k-mer té moltes implicacions en l'assemblatge de seqüències. Aquests efectes varien molt entre aquells k-mers de mida major i menor. A continuació es descriuen alguns dels efectes segons les mides.
k-mers de mides menors
- Un k-mer de mida menor disminueix la quantitat d'arestes emmagatzemades en el graf, i com a tal, ajudarà a disminuir la quantitat d'espai necessari per a emmagatzemar les seqüències d'ADN.
- Les mides menors augmenten la possibilitat que tots el k-mers se solapin, i com a tal, tenen així les subseqüències suficients per a construir el graf de Bruijn.[4]
- Tanmateix, en tenir mides de k-mers més petites, també hi ha el risc de tenir més vèrtexs en el graf que dirigeixen a un sol k-mer. Per això, això farà la reconstrucció del genoma més difícil mentre hi hagi un nivell més alt d'ambigüitats de camí a causa de la major quantitat vèrtexs que cal que es recorrin.
- La informació es perd a mesura que el k-mers són menors.
- P. ex., la possibilitat de AGTCGTAGATGCTG és menor que ACGT, i com a tal, conté una quantitat major d'informació (vegeu entropia de Shannon).
- Els k-mers més petits també tenen el problema de no ser capaç de resoldre àrees a l'ADN on hi hagi microsatèl·lits o repeticions. Això és perquè els k-mers més petits tendiran a caure enterament dins d'una regió repetitiva i per això es difícil determinar la quantitat de repetició que ha tingut lloc.
- P. ex., per a la subseqüència ATGTGTGTGTGTGTACG, la quantitat de les repeticions de TG es perdran si es tria un k-mer de mida menor de 16. Això és perquè la majoria del k-mers cauran en la regió repetida i només pot ser descartada mentre repeteixi el mateix k-mer en comptes de referir-se la quantitat de repeticions.
k-mers de mides majors
- Amb k-mers de mides majors s'augmentarà la quantitat d'arestes en el graf, que implicarà un augment de la quantitat de memòria necessària per emmagatzemar la seqüència d'ADN.
- En augmentar la mida del k-mers, el nombre de vèrtexs també disminuirà. Això ajudarà la construcció del genoma perquè hi hi haurà menys camins per recórrer en el graf.[5]
- Els k-mers de mides majors corren risc el risc de no tenir vèrtexs de fora de cada k-mer. Això és perquè els k-mers majors augmenten el risc que no se solapin amb un altre k-mer per . Això pot portar a desunions en les lectures, i a un major nombre de contigs menors.
- Els k-mers alleugen el problema de regions repetitives més petites. Això és perquè el k-mer contindrà un equilibri de regions repetitives i seqüències d'ADN adjuntes (donat que siguin d'una mida prou gran) que pot ajudar a resoldre la quantitat de repetició en aquella àrea particular.
Aplicacions dels k-mers en les anàlisis bioinformàtiques
La freqüència d'un conjunt de k-mers en el genoma d'una espècie, en una regió genòmica, o en una classe de seqüències, pot ser utilitzat com a «signatura» de la seqüència subjacent. Comparar aquestes freqüències és computacionalment més fàcil que un alineament de seqüència, i és un mètode important en l'anàlisi de seqüències sense alineaments. També pot utilitzar-se com a primer estadi d'anàlisi abans d'un alineament.
- Separació d'espècies diferents en una mescla de material genètic (metagenòmica, microbioma);[6][7] pot afegir-se informació de fase/marc de lectura[8]
- Barcoding d'ADN d'espècies[9][10]
- Assemblatge de seqüències de novo[11]
- Classificació de l'haplogrup mitocondrial humà[12]
- Detecció d'assemblatge incorrecte de genoma[13]
- Detecció de novo de seqüències repetides com ara elements transposables[14]
- Caracterització d'un motiu de seqüència d'unió a proteïnes.[15] A més dels k-mer, també es poden utilitzar k-mers amb buits (també anomenats q-grams amb buits o llavors espaiades)[16]
- Identificació de mutacions o polimorfismes utilitzant dades de sequenciació[17]
- Caracterització d'illes CpG per regions flanquejants
- Detecció de transferència horitzontal
- Detecció de contaminació bacteriana en l'assemblatge d'un genoma eucariota
- Detecció de llocs de recombinació
- Ús de la freqüència del k-mer respecte la profunditat del k-mer per calcular la mida de genoma
- Estimació del nivell de RNA-seq
Pseudocodi
Determinar els possible k-mers d'una lectura pot fer-se simplement entrant en un bucle prenent la longitud de la cadena per un i prenent cada subcadena de longitud, k. El pseudocodi per a fer-ho podria ser el següent:
procediment k-mer(Cadena, k : longitud de cada k-mer) n = longitud(Cadena) /* itera sobre la longitud de la Cadena fins que es puguin fer k-mers de longitud k */ itera i = 1 a n-k+1 inclosos fes /* sortida de cada k-mer de longitud k, de i a i+k en Cadena*/ sortida Cadena[i:i+k] fi itera fi procediment
Python
def find_kmers(string, k): kmers = [] n = len(string) for i in range(0, n-k+1): kmers.append(string[i:i+k]) return kmers
Exemples
A continuació es mostren alguns exemples amb els possibles k-mers de seqüències d'ADN (donat un valor de k determinat):
Seq: AGATCGAGTG 3-mers: AGA GAT ATC TCG CGA GAG AGT GTG
Seq: GTAGAGCTGT 5-mers: GTAGA TAGAG AGAGC GAGCT AGCTG GCTGT
Referències
- ↑ 1,0 1,1 Compeau, Phillip E C; Pevzner, Pavel A; Tesler, Glenn «How to apply de Bruijn graphs to genome assembly» (en anglès). Nature Biotechnology, 29, 11, 2011/11, pàg. 987–991. DOI: 10.1038/nbt.2023. ISSN: 1546-1696.
- ↑ Samocha, Kaitlin E; Robinson, Elise B; Sanders, Stephan J; Stevens, Christine; Sabo, Aniko; McGrath, Lauren M; Kosmicki, Jack A; Rehnström, Karola; Mallick, Swapan «A framework for the interpretation of de novo mutation in human disease». Nature Genetics, 46, 9, 2014, pàg. 944–950. DOI: 10.1038/ng.3050. ISSN: 1061-4036.
- ↑ Aggarwala, Varun; Voight, Benjamin F «An expanded sequence context model broadly explains variability in polymorphism levels across the human genome». Nature Genetics, 48, 4, 2016, pàg. 349–355. DOI: 10.1038/ng.3511. ISSN: 1061-4036. PMC: 4811712. PMID: 26878723.
- ↑ Zerbino, Daniel R.; Birney, Ewan «Velvet: Algorithms for de novo short read assembly using de Bruijn graphs». Genome Research, 18, 5, 2008-5, pàg. 821–829. DOI: 10.1101/gr.074492.107. ISSN: 1088-9051. PMC: PMC2336801. PMID: 18349386.
- ↑ Zerbino, Daniel R.; Birney, Ewan «Velvet: Algorithms for de novo short read assembly using de Bruijn graphs» (en anglès). Genome Research, 18, 5, 01-05-2008, pàg. 821–829. DOI: 10.1101/gr.074492.107. ISSN: 1088-9051. PMID: 18349386.
- ↑ Ounit, Rachid; Wanamaker, Steve; Close, Timothy J.; Lonardi, Stefano «CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers». BMC Genomics, 16, 25-03-2015, pàg. 236. DOI: 10.1186/s12864-015-1419-2. ISSN: 1471-2164.
- ↑ Dubinkina, Veronika B.; Ischenko, Dmitry S.; Ulyantsev, Vladimir I.; Tyakht, Alexander V.; Alexeev, Dmitry G. «Assessment of k-mer spectrum applicability for metagenomic dissimilarity analysis». BMC Bioinformatics, 17, 16-01-2016, pàg. 38. DOI: 10.1186/s12859-015-0875-7. ISSN: 1471-2105.
- ↑ Zhu, Jianfeng; Zheng, Wei-Mou «Self-organizing approach for meta-genomes». Computational Biology and Chemistry, 53, Part A, 01-12-2014, pàg. 118–124. DOI: 10.1016/j.compbiolchem.2014.08.016.
- ↑ Chor, Benny; Horn, David; Goldman, Nick; Levy, Yaron; Massingham, Tim «Genomic DNA k-mer spectra: models and modalities». Genome Biology, 10, 08-10-2009, pàg. R108. DOI: 10.1186/gb-2009-10-10-r108. ISSN: 1474-760X.
- ↑ Meher, Prabina Kumar; Sahu, Tanmaya Kumar; Rao, A. R. «Identification of species based on DNA barcode using k-mer feature vector and Random forest classifier». Gene, 592, 2, 05-11-2016, pàg. 316–324. DOI: 10.1016/j.gene.2016.07.010.
- ↑ Li, Ruiqiang; Zhu, Hongmei; Ruan, Jue; Qian, Wubin; Fang, Xiaodong «De novo assembly of human genomes with massively parallel short read sequencing». Genome Research, 20, 2, 2010-2, pàg. 265–272. DOI: 10.1101/gr.097261.109. ISSN: 1088-9051. PMC: PMC2813482. PMID: 20019144.
- ↑
- ↑ Phillippy, Adam M.; Schatz, Michael C.; Pop, Mihai «Genome assembly forensics: finding the elusive mis-assembly». Genome Biology, 9, 14-03-2008, pàg. R55. DOI: 10.1186/gb-2008-9-3-r55. ISSN: 1474-760X.
- ↑ Price, A. L.; Jones, N. C.; Pevzner, P. A. «De novo identification of repeat families in large genomes» (en anglès). Bioinformatics, 21, Suppl 1, 01-06-2005, pàg. i351–i358. DOI: 10.1093/bioinformatics/bti1018. ISSN: 1367-4803.
- ↑ Newburger, D. E.; Bulyk, M. L. «UniPROBE: an online database of protein binding microarray data on protein-DNA interactions» (en anglès). Nucleic Acids Research, 37, Database, 01-01-2009, pàg. D77–D82. DOI: 10.1093/nar/gkn660. ISSN: 0305-1048.
- ↑ Ghandi, Mahmoud; Lee, Dongwon; Mohammad-Noori, Morteza; Beer, Michael A. «Enhanced Regulatory Sequence Prediction Using Gapped k-mer Features». PLOS Computational Biology, 10, 7, 17-07-2014, pàg. e1003711. DOI: 10.1371/journal.pcbi.1003711. ISSN: 1553-7358.
- ↑ Nordström, Karl J V; Albani, Maria C; James, Geo Velikkakam; Gutjahr, Caroline; Hartwig, Benjamin «Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers» (en anglès). Nature Biotechnology, 31, 4, 2013/04, pàg. 325–330. DOI: 10.1038/nbt.2515. ISSN: 1546-1696.